library(tidyverse)── Attaching core tidyverse packages ──────────────────────── tidyverse 2.0.0 ──
✔ dplyr 1.2.1 ✔ readr 2.2.0
✔ forcats 1.0.1 ✔ stringr 1.6.0
✔ ggplot2 4.0.3 ✔ tibble 3.3.1
✔ lubridate 1.9.5 ✔ tidyr 1.3.2
✔ purrr 1.2.2
── Conflicts ────────────────────────────────────────── tidyverse_conflicts() ──
✖ dplyr::filter() masks stats::filter()
✖ dplyr::lag() masks stats::lag()
ℹ Use the conflicted package (<http://conflicted.r-lib.org/>) to force all conflicts to become errorslibrary(faraway)
library(patchwork)
data("gala")
glimpse(gala)Rows: 30
Columns: 7
$ Species <dbl> 58, 31, 3, 25, 2, 18, 24, 10, 8, 2, 97, 93, 58, 5, 40, 347, …
$ Endemics <dbl> 23, 21, 3, 9, 1, 11, 0, 7, 4, 2, 26, 35, 17, 4, 19, 89, 23, …
$ Area <dbl> 25.09, 1.24, 0.21, 0.10, 0.05, 0.34, 0.08, 2.33, 0.03, 0.18,…
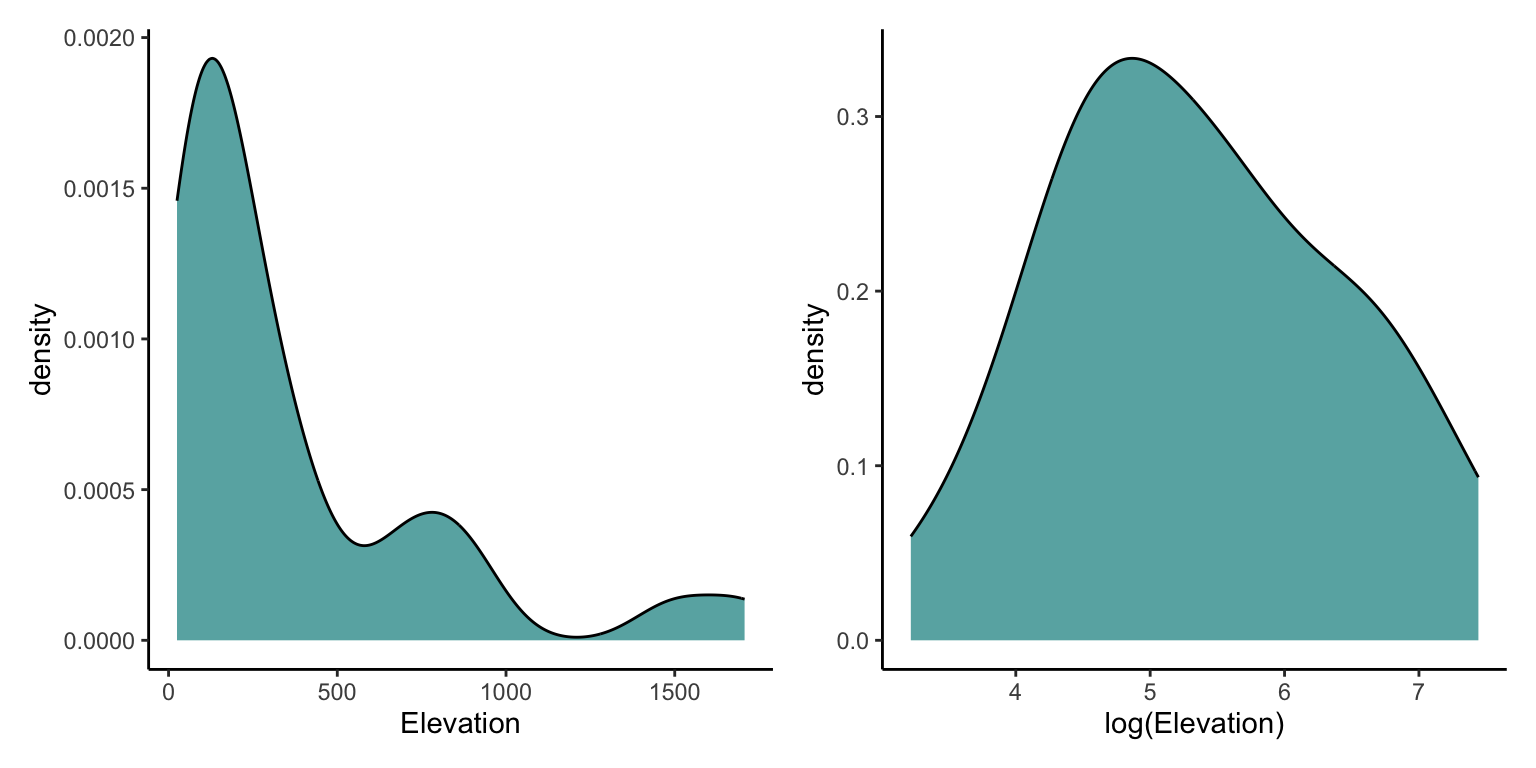
$ Elevation <dbl> 346, 109, 114, 46, 77, 119, 93, 168, 71, 112, 198, 1494, 49,…
$ Nearest <dbl> 0.6, 0.6, 2.8, 1.9, 1.9, 8.0, 6.0, 34.1, 0.4, 2.6, 1.1, 4.3,…
$ Scruz <dbl> 0.6, 26.3, 58.7, 47.4, 1.9, 8.0, 12.0, 290.2, 0.4, 50.2, 88.…
$ Adjacent <dbl> 1.84, 572.33, 0.78, 0.18, 903.82, 1.84, 0.34, 2.85, 17.95, 0…